Here is some code to run Phylometa from R. Phylometa is a program that conducts phylogenetic meta-analyses. The great advantage of the approach below is that you can easily run Phylometa from R, and manipulate the output from Phylometa in R.
Phylometa was created by Marc Lajeunesse at University of South Florida, and is described in his 2009 AmNat paper. Phylometa can be downloaded free here.
Save phylometa_fxn.R (get here) to your working directory. Then use the block of code below to call the functions within phylometa_fxn.R.
The program Phylometa needs to be in the working directory you are calling from.
Let me know what doesn’t work, and what improvements can be made; I’m sure there are many!
—This code below is also available here on Github.
########Directions
#Place phylometa software to your working directory
#Put your phylogeny, in format required by phylometa, in your working directory
#Put your meta-analysis dataset, in format required by phylometa, in your working directory
#Set working directory
#Use below functions
#Beware: only use a moderator variable with up to 6 groups
########Install packages
install.packages(c("plyr","ggplot2"))
library(plyr)
library(ggplot2)
########Set the working directory [NOTE:CHANGE TO YOUR WORKING DIRECTORY]
setwd("/Users/Scott/Documents/phylometa")
#Call and run functions (used below) in the working directory [NOTE:CHANGE TO YOUR WORKING DIRECTORY]
source("/Users/Scott/Documents/phylometa")
###########################Functions to to a phylogenetic meta-analysis
#Define number of groups in moderator variable
groups <- 2
####Run phylometa. Change file names as needed
phylometa.run <- system(paste('"phyloMeta_v1-2_beta.exe" phylogeny.txt metadata_2g.txt'),intern=T)
####Process phylometa output
#E.g.
myoutput <- phylometa.process(phylometa.run,groups)
####Get output from phylometa.run
phylometa.output(myoutput) #Prints all five tables
phylometa.output.table(myoutput,2) #Prints the table you specify, from 1 to 5, in this example, table 2 is output
###################################################
#########Plot effect sizes. These are various ways to look at the data. Go through them to see what they do. Output pdf's are in your working directory
#Make table for plotting
analysis <- c(rep("fixed",groups+1),rep("random",groups+1))
trad_effsizes <- data.frame(analysis,phylometa.output.table(myoutput,2)) #Tradiational effect size table
phylog_effsizes <- data.frame(analysis,phylometa.output.table(myoutput,4)) #Phylogenetic effect size table
#The arrange method
limits <- aes(ymax = effsize + (CI_high-effsize), ymin = effsize - (effsize-CI_low))
dodge <- position_dodge(width=0.3)
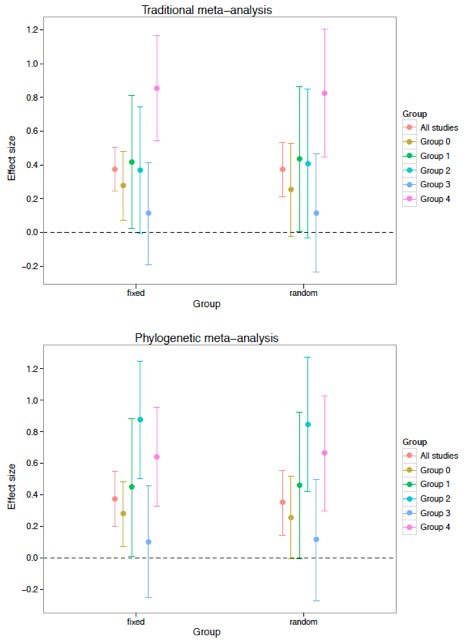
plot01 <- ggplot(trad_effsizes,aes(y=effsize,x=analysis,colour=Group)) + geom_point(size=3,position=dodge) + theme_bw() + opts(panel.grid.major = theme_blank(),panel.grid.minor=theme_blank(),title="Traditional meta-analysis") + labs(x="Group",y="Effect size") + geom_errorbar(limits, width=0.2, position=dodge) + geom_hline(yintercept=0,linetype=2)
plot02 <- ggplot(phylog_effsizes,aes(y=effsize,x=analysis,colour=Group)) + geom_point(size=3,position=dodge) + theme_bw() + opts(panel.grid.major = theme_blank(),panel.grid.minor=theme_blank(),title="Phylogenetic meta-analysis") + labs(x="Group",y="Effect size") + geom_errorbar(limits, width=0.2, position=dodge) + geom_hline(yintercept=0,linetype=2)
pdf("plots_effsizes_arrange.pdf",width = 8, height = 11)
arrange(plot01,plot02,ncol=1)
dev.off()
#used in the two plotting methods below
bothanalyses<-data.frame(tradphy=c(rep("Traditional",(groups*2)+2),rep("Phylogenetic",(groups*2)+2)),fixrand=rep(analysis,2),rbind.fill(phylometa.output.table(myoutput,2),phylometa.output.table(myoutput,4))) #Table of both trad and phylo
limits2 <- aes(ymax = effsize + (CI_high-effsize), ymin = effsize - (effsize-CI_low))
dodge <- position_dodge(width=0.3)
#The grid/wrap method, version 1
plot03 <- ggplot(bothanalyses,aes(y=effsize,x=tradphy,colour=Group)) + geom_point(size=3,position=dodge) + theme_bw() + opts(panel.grid.major = theme_blank(),panel.grid.minor=theme_blank()) + labs(x="Group",y="Effect size") + geom_errorbar(limits2, width=0.2, position=dodge) + geom_hline(yintercept=0,linetype=2) + facet_grid(.~fixrand)
pdf("plots_effsizes_wrap1.pdf")
plot03
dev.off()
#The grid/wrap method, version 2 (excuse the sloppy x-axis labels)
plot04 <- ggplot(bothanalyses,aes(y=effsize,x=Group,colour=tradphy)) + geom_point(size=3,position=dodge) + theme_bw() + opts(panel.grid.major = theme_blank(),panel.grid.minor=theme_blank()) + labs(x="Group",y="Effect size") + geom_errorbar(limits2, width=0.2, position=dodge) + geom_hline(yintercept=0,linetype=2) + facet_grid(.~fixrand)
pdf("plots_effsizes_wrap2.pdf")
plot04
dev.off()
Below is an example output figure from the code. This example is from an analysis using 5 groups (i.e., 5 levels in the explanatory variable).